|
Introduction
This extended tutorial is made up of five tutorials designed especially for biomedical researchers so that they can get a basic understanding of the functions of Vect for data extraction and conversion. Users should be able to perform the following tasks upon completion of the tutorial;
- Load the file to be processed
- Select regions you want to use
- Apply rules to the data set
- Arrange the data in a desired format
- Convert the final format to programming code
Download the Arabidopsis file used in this example by clicking here here.
(Alternatively you can obtain it by going to the GenBank Homepage < http://www.ncbi.nlm.nih.gov> and searching for the 'AC006439' file under the 'Nucleotide' search. Click on the AC006439 hyperlink to open the file. Select 'Text,' click on 'Send to' and save the file as AC006439.txt by selecting File > Save As and changing the pull down menu to 'Text File.')
Open Vect and open the AC006439.txt file through the 'Open' icon or select Files > Open files from the pull down menu. The file should appear in the body. Use the scroll bar to view the whole file. The number at the left most side represent line numbers (1, 2, 3, …) and the other numbers (1, 1, 1, …) besides the line numbers represent the level of data.
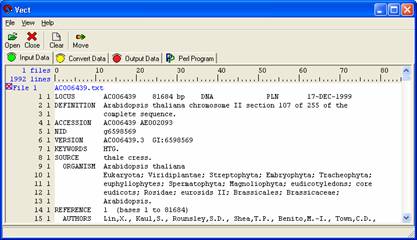
At the completion of this tutorial, the following items will be extracted from the original document. Each number refers to a short tutorial within this document;
1) gene sequence (end of file)
2) protein id (/protein_id=)
3) gene names (/gene=)
4) mRNA join coordinates (/gene=)
5) protein sequence (/translation=)
During the final stages, this data that the user extracted will be arranged in a way the user wants to format. Then a Perl program will be generated such that the Perl program will provide similar results for all semi-structured data in the nucleotide search under the GenBank Homepage.

|

